Le tronc artériel commun est une cardiopathie conotroncale dans laquelle une artère unique assure l'éjection du sang venant à la fois du ventricule gauche et du ventricule droit. Ce tronc artériel unique émerge de la base du coeur au-dessus d'une valve semi-lunaire unique, appelée valve troncale, et donne naissance aux artères coronaires, aux artères pulmonaires, et à l'aorte (Figure).

C’est une malformation rare avec une incidence de 0,04 pour 1000 naissances vivantes [1]. Elle représente 0,3 % des cardiopathies congénitales [2].
L’association à un syndrome microdélétionnel 22q11 est fréquente et doit être systématiquement recherchée.
La variabilité de l’origine des branches pulmonaires est la base de nombreuses classifications anatomiques dont la plus ancienne est celle de Collett et Edwards. Dans le type I, les artères pulmonaires proviennent d’un tronc artériel pulmonaire (c’est le cas de notre patient).
Dans le type II, elles se détachent directement du TAC, proches l’une de l’autre, contrairement au type III où elles sont éloignées l’une de l’autre.
Dans le type IV, des artères collatérales de l’aorte descendante vascularisent les poumons et les branches pulmonaires sont absentes (pseudotruncus arteriosus).

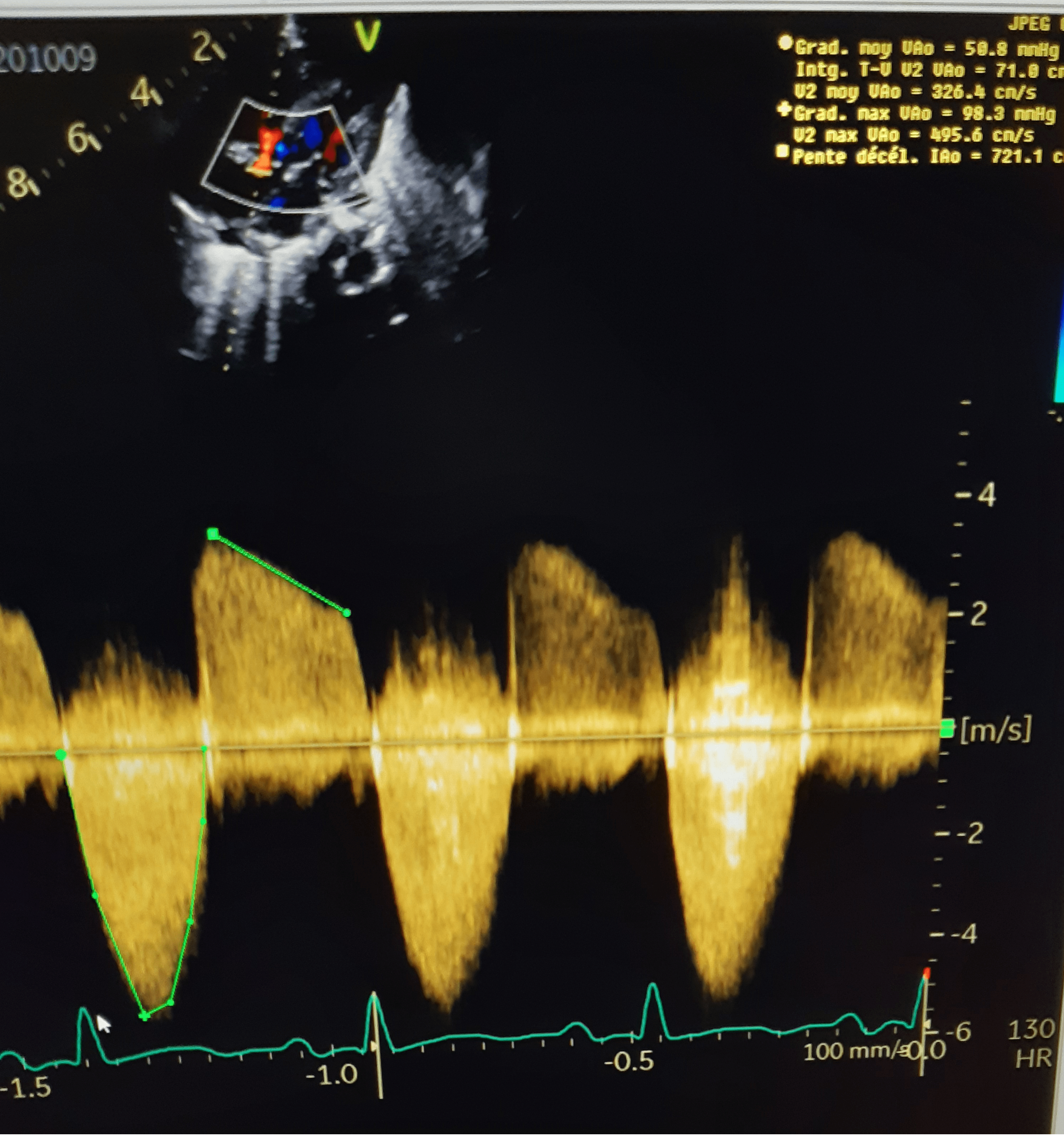
La valve troncale a un nombre variable de feuillets. Elle peut être tricuspide, quadricuspide ou bicuspide dans respectivement 69, 21, et 9 % des cas. La valve peut être également très dysplasique conduisant à une insuffisance dans près de 50 % des cas, et/ou à une sténose chez près de 20 % des patients [3].
Le cas présenté illustre une valve fortement dysplasique avec fuite et sténose significatives, ce qui aggrave notablement le pronostic. En effet, c’est une cardiopathie caractérisée sur le plan physiopathologique par un mélange complet des retours veineux cave et pulmonaire par un shunt bidirectionnel au niveau de la CIV, avant la sortie par le truncus arteriosus qui est la seule voie d’évacuation des deux ventricules.
L’évolution spontanée sans traitement est sombre puisqu’elle se fait en général soit vers une insuffisance cardiaque néonatale congestive précipitée dans notre cas par l’hypertrophie ventriculaire gauche importante sur sténsoe valvulaire sévère, soit, sans traitement, vers une maladie vasculaire pulmonaire obstructive, parfois avant l’âge de 1 an.
Au cours du premier mois de vie , et avec baisse de résistances vasculaires pulmonaires , il y a un hyper débit pulmonaire et un risque de vol coronaire et par conséquent d’ischémie myocardique. Cette ischémie a été également rapide chez notre patient survenue à j3 de vie car les coronaires prennent naissance dans une valve très remaniée épaissie responsable d’un vrai hypo-débit coronaire.
Le traitement définitif ne peut être que chirurgical, et consiste à fermer la communication interventriculaire, désinsérer les artères pulmonaires du truncus et rétablir la continuité entre le ventricule droit et les branches artérielles
pulmonaires par une homogreffe valvée,avec plastie de la valve troncale dans les meilleurs cas et ceci dans les 3 premiers mois de vie ; des réopérations multiples sont nécessaires par la suite pour changer la voie pulmonaire, pour des raisons d’absence de croissance ou de sténose.
Pour notre patient , l’atteinte sévère de la valve troncale a aboutit en plus au remplacement de la voie gauche par une homogreffe aortique avec réimplantation des artères coronaires .Le résultat avec un recul de 3 mois est satisfaisant.
Références:
1/ Freedom RM. Anomalies of aortopulmonary septation: persistent truncus arteriosus, aortopulmonary septal defect and hemitruncus arteriosus. Neonatal heat disease. London: Springer Verlag; 1992. p. 425-52.
2/ Keane JF, Fyler DC. Truncus arteriosus.NADAS’ Pediatric cardiology. Philadelphia: WB Saunders-Elsevier; 2006. p. 767-71.
3/ Butto F, Lucas RV, Edwards JE. Persistent truncus arteriosus:
pathologic anatomy in 54 cases. Pediatr Cardiol 1986;7:95-101.
4. O. Raisky, P. Vouhé. Tronc artériel commun.Traitement chirurgical. Traité d’EMC. Techniques chirurgicales – Thorax 2007.